
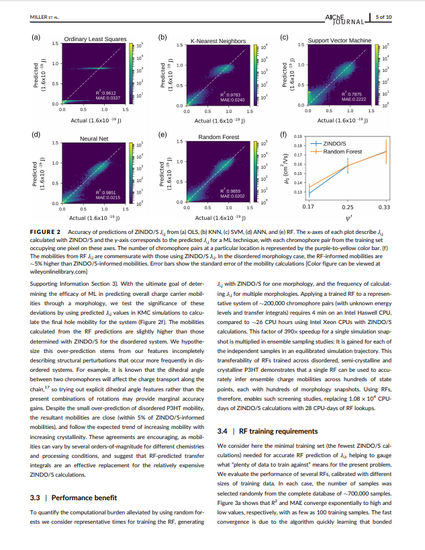
The purpose of this work is to lower the computational cost of predicting charge mobilities in organic semiconductors, which will benefit the screening of candidates for inexpensive solar power generation. We characterize efforts to minimize the number of expensive quantum chemical calculations we perform by training machines to predict electronic couplings between monomers of poly-(3-hexylthiophene). We test five machine learning techniques and identify random forests as the most accurate, information-dense, and easy-to-implement approach for this problem, achieving mean-absolute-error of 0.02 [× 1.6 × 10−19 J], R2 = 0.986, predicting electronic couplings 390 times faster than quantum chemical calculations, and informing zero-field hole mobilities within 5% of prior work. We discuss strategies for identifying small effective training sets. In sum, we demonstrate an example problem where machine learning techniques provide an effective reduction in computational costs while helping to understand underlying structure–property relationships in a materials system with broad applicability.
Available at: http://works.bepress.com/eric_jankowski/40/